Un po' di storia
Alcuni ricordi storici possono servire per meglio valutare sia la situazione attuale
negli animali che il rischio di trasmissione all'uomo, nella forma identificata
come
variante del morbo di Creutzfeldt-Jacob (vCJD)![]() .
.
 |
|
 |


 |
|
Clicca sull'immagine per vedere il filmato |
La prima malattia da prioni![]() riconosciuta è stata descritta nelle
pecore con il termine scrapie (1732). La denominazione si deve al prurito
che gli animali colpiti manifestano, da to scrape: grattarsi. Tuttavia
già Dante nel quinto canto del Paradiso afferma "siate uomini non pecore matte",
ricordando così una sintomatologia che poteva essere riportata a forme sostenute
da agenti parassitari ma anche alla scrapie, diagnosticata come malattia
molti secoli dopo.
riconosciuta è stata descritta nelle
pecore con il termine scrapie (1732). La denominazione si deve al prurito
che gli animali colpiti manifestano, da to scrape: grattarsi. Tuttavia
già Dante nel quinto canto del Paradiso afferma "siate uomini non pecore matte",
ricordando così una sintomatologia che poteva essere riportata a forme sostenute
da agenti parassitari ma anche alla scrapie, diagnosticata come malattia
molti secoli dopo.
In Francia nel 1883 viene segnalato uno strano caso di
scrapie osservato in una bovina nel 1881. In seguito le encefalopatie
spongiformi trasmissibili (TSE) sono state segnalate in diverse specie animali
quali bovini
(Bovine Spongiform Encefhalopaty, BSE), felini
(Feline Spongiform Encephalopaty, FSE![]() ), visoni
(Transmissible Mink Encephalopaty, TME
), visoni
(Transmissible Mink Encephalopaty, TME![]() ), ruminanti selvatici quali il
cervo come malattia cronica devastante
(Chronic Wasting Disease, CWD
), ruminanti selvatici quali il
cervo come malattia cronica devastante
(Chronic Wasting Disease, CWD![]() ) e in animali esotici degli zoo
quali ocelot, puma, tigre, leone, bisonte, antilope camoscio (Oryx gazella)
e antilope alcina (Taurotragus oryx).
) e in animali esotici degli zoo
quali ocelot, puma, tigre, leone, bisonte, antilope camoscio (Oryx gazella)
e antilope alcina (Taurotragus oryx).
 |
 |
 |
Una delle forme segnalate nell'uomo e riconosciute ad eziologia![]() prionica
è il Kuru
prionica
è il Kuru![]() ,
epidemico in Nuova Guinea, trasmesso dalle pratiche legate al cannibalismo. Dovrebbe essere cessato nel 1958 circa.
È interessante rilevare come donne e bambini che
partecipavano al rito manipolando gli organi che sarebbero stati consumati dagli
uomini adulti, presentavano più frequentemente i segni della malattia. In base a
tale evidenza, sembrerebbe che la malattia si trasmettesse più per contatto
diretto con gli organi infetti che per ingestione di questi. D'altro canto, non
si può neppure escludere l'ipotesi che donne e bambini assaggiassero di nascosto quanto
preparavano.
,
epidemico in Nuova Guinea, trasmesso dalle pratiche legate al cannibalismo. Dovrebbe essere cessato nel 1958 circa.
È interessante rilevare come donne e bambini che
partecipavano al rito manipolando gli organi che sarebbero stati consumati dagli
uomini adulti, presentavano più frequentemente i segni della malattia. In base a
tale evidenza, sembrerebbe che la malattia si trasmettesse più per contatto
diretto con gli organi infetti che per ingestione di questi. D'altro canto, non
si può neppure escludere l'ipotesi che donne e bambini assaggiassero di nascosto quanto
preparavano.
Altre forme di encefalopatia nell'uomo sono: la sindrome Creutzfeldt-Jacob (CJD)![]() ,
nella forma sporadica, ereditaria e iatrogena
,
nella forma sporadica, ereditaria e iatrogena![]() , la variante del morbo di
Creutzfeldt-Jakob (vCJD), la sindrome Gerstmann-Sträussler-Scheinker (GSS)
, la variante del morbo di
Creutzfeldt-Jakob (vCJD), la sindrome Gerstmann-Sträussler-Scheinker (GSS)![]() e
l'insonnia fatale familiare (FFI)
e
l'insonnia fatale familiare (FFI)![]() .
.
Clinicamente queste malattie decorrono con perdita progressiva nella
coordinazione dei movimenti e deterioramento intellettivo che porta
inevitabilmente alla demenza. Il periodo d'incubazione è lungo, il decorso è
cronico e per ora non esiste terapia. Nei soggetti colpiti non si riscontrano
lesioni infiammatorie né risposte immunitarie. Vi sono, ma non sempre, lesioni
degenerative a carattere spongioso nel sistema nervoso centrale. In
particolare l'accumulo di vacuoli![]() intracellulari si ha nella CJD, mentre prevale
la formazione di fibrille di amiloide
intracellulari si ha nella CJD, mentre prevale
la formazione di fibrille di amiloide![]() nella GSS e l'abiotrofia
nella GSS e l'abiotrofia![]() talamica,
scomparsa delle cellule nervose del talamo, caratterizza la FFI. È presente
comunque sempre una proteina patologica specifica, prione, denominata PrPsc
(sc=scrapie) o PrPres (res=resistente alle proteasi
talamica,
scomparsa delle cellule nervose del talamo, caratterizza la FFI. È presente
comunque sempre una proteina patologica specifica, prione, denominata PrPsc
(sc=scrapie) o PrPres (res=resistente alle proteasi![]() ).
Questa proteina patologica, probabilmente causa della sintomatologia, tende a
modificare la proteina prionica PrP, normalmente presente nell'organismo,
denominata PrPsen (sen=sensibile alle proteasi) o PrPc (c=cellulare) della quale ancora non è nota
la funzione.
).
Questa proteina patologica, probabilmente causa della sintomatologia, tende a
modificare la proteina prionica PrP, normalmente presente nell'organismo,
denominata PrPsen (sen=sensibile alle proteasi) o PrPc (c=cellulare) della quale ancora non è nota
la funzione.
|
|
|
|
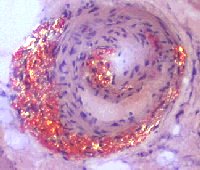
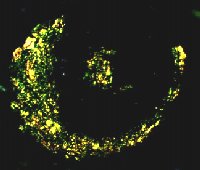
Fig. 1: Amiloide evidenziata con la colorazione Rosso Congo (a sinistra) e in luce polarizzata (a destra). |
|