Il cervello: dal tessuto normale alla malattia
Il Sistema Nervoso Centrale
Il sistema nervoso centrale (SNC) è suddiviso in due parti principali: il cervello ed il midollo spinale. Nell'uomo adulto, il cervello pesa mediamente da 1,3 a 1,4 Kg.
 Fig. 1: Il cervello |
Il cervello, inteso come la porzione di SNC contenuta all'interno della scatola cranica, contiene circa 100 bilioni di cellule nervose (neuroni![]() ) e trilioni di "cellule di supporto, chiamate, nel complesso, glia
) e trilioni di "cellule di supporto, chiamate, nel complesso, glia![]() . In questo ammasso di cellule, altamente organizzato, vengono generate molte delle funzioni, consce o inconsce, indispensabili per la nostra vita, dall'apprendimento al movimento, dal linguaggio alle emozioni, fino alla regolazione del funzionamento di altri organi, come il battito cardiaco o la respirazione.
. In questo ammasso di cellule, altamente organizzato, vengono generate molte delle funzioni, consce o inconsce, indispensabili per la nostra vita, dall'apprendimento al movimento, dal linguaggio alle emozioni, fino alla regolazione del funzionamento di altri organi, come il battito cardiaco o la respirazione.
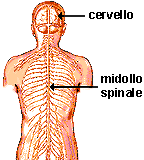 Fig. 2: rappresentazione della localizzazione del cervello e midollo spinale |
Come descritto in http://www.scienzagiovane.unibo.it/Cervello.html, il cervello è un sistema altamente complesso e finemente regolato e, di conseguenza, tutti i fattori che in qualche modo alterino questa organizzazione, possono essere causa di degenerazione nervosa.
Quali meccanismi alla base delle malattie neurodegenerative?
Possiamo distinguere, in base alla natura dell'evento responsabile dell'insorgenza di una malattia neurodegenerativa, diversi meccanismi che, pur derivando da cause diverse, possono molto spesso essere compresenti durante la progressione e l’evoluzione di un quadro patologico.
Come molte altre patologie, anche le malattie neurodegenerative possono essere causate dalla presenza di cosiddette mutazioni![]() in alcuni geni
in alcuni geni![]() presenti nel nostro DNA. L'effetto di una mutazione su un gene spesso può avere l'effetto di portare alla mancata produzione di una proteina, oppure generare proteine alterate, malfunzionanti o persino tossiche per la cellula. L'effetto finale di queste modificazioni dipende dal gene interessato e dal tipo di cellula in cui esse si verificano.
presenti nel nostro DNA. L'effetto di una mutazione su un gene spesso può avere l'effetto di portare alla mancata produzione di una proteina, oppure generare proteine alterate, malfunzionanti o persino tossiche per la cellula. L'effetto finale di queste modificazioni dipende dal gene interessato e dal tipo di cellula in cui esse si verificano.
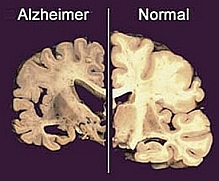 Fig. 3: Differenze anatomiche tra sezioni di cervello: a sinistra un cervello affetto da Alzheimer, a destra uno normale. |
Nelle malattie neurodegenerative, in particolare, mutazioni su geni funzionalmente diversi, spesso portano allo stesso profilo di degenerazione. È quanto succede quando la proteina alterata perde la sua normale conformazione e tende ad aggregarsi; questo dà origine a fibrille o placche intra- e/o extracellulari che, col passare del tempo si accrescono portando alla morte della cellula e del tessuto in cui questi aggregati si formano.
È il caso delle cosiddette “malattie da ripetizioni di triplette”, un set di disordini genetici causati dall'allungamento di ripetizioni di trinucleotidi![]() in alcuni geni. Alcune di queste ripetizioni, infatti, rappresentano delle regioni altamente instabili del genoma che, a causa di errori durante i processi di replicazione del DNA, tendono ad aumentare di numero. Al di sotto di una certa soglia, che differisce dal tipo di gene, queste ripetizioni non hanno effetto sulla funzione della proteina codificata. Quando però il numero di triplette supera il valore limite, la proteina acquisisce una funzione tossica per la cellula, in quanto tende ad aggregare.
in alcuni geni. Alcune di queste ripetizioni, infatti, rappresentano delle regioni altamente instabili del genoma che, a causa di errori durante i processi di replicazione del DNA, tendono ad aumentare di numero. Al di sotto di una certa soglia, che differisce dal tipo di gene, queste ripetizioni non hanno effetto sulla funzione della proteina codificata. Quando però il numero di triplette supera il valore limite, la proteina acquisisce una funzione tossica per la cellula, in quanto tende ad aggregare.
La malattia di Huntington è forse l'esempio più tipico di questa classe di malattie. Qui, infatti, la causa è rappresentata da un eccesso di ripetizioni CAG nel gene dell'Huntingtina (HTT), che codificano per l’aminoacido Glutamina. La maggior parte degli individui sani hanno un numero di triplette in questo gene inferiore a 28. Nei soggetti in cui, per i meccanismi sopra descritti viene superato il numero di 36 ripetizioni, la proteina tende a formare aggregati responsabili della manifestazione della malattia.
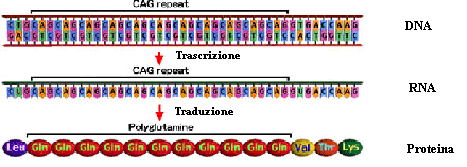
Fig. 4: La alterazione del numero delle triplette di nucleotidi CAG nel DNA si riflette, tramite i meccanismi di trascrizione in RNA e codificazione in aminoacidi, nellaumento del numero di glutamine nella proteina
Meccanismi comuni sono causa di alcune forme di Atassia![]() spino-cerebellare o della sindrome dell'X fragile.
spino-cerebellare o della sindrome dell'X fragile.
Aggregati proteici possono formarsi anche in seguito a sostituzioni di un singolo aminoacido nella proteina come nel gene per l'alfa-sinucleina in alcuni casi di malattia di Parkinson o di Alzheimer.
Un altro caso molto particolare è quello della Sindrome della Mucca Pazza, descritto ampiamente in http://www.scienzagiovane.unibo.it/muccapazza.html.
Questa sindrome, che scientificamente viene chiamata nell'uomo “Sindrome di Creutzfeld-Jacob”, rappresenta la prima malattia neurodegenerativa veicolata all'uomo tramite cibo. In particolare, esiste una proteina espressa nel cervello dei bovini (molto simile a quella prodotta nell'uomo), il prione![]() , che, nell'encefalopatia spongiforme bovina, risulta essere mutata e particolarmente resistente anche alla cottura. Se attraverso la carne questa proteina viene introdotta nell'uomo, tende ad accumularsi in alcune regioni del cervello senza poter essere distrutta. Inoltre la proteina alterata può portare addirittura all'alterazione delle proteine prioniche normali prodotte dalle nostre cellule, innescando così, un meccanismo a catena con accumulo di prioni alterati.
, che, nell'encefalopatia spongiforme bovina, risulta essere mutata e particolarmente resistente anche alla cottura. Se attraverso la carne questa proteina viene introdotta nell'uomo, tende ad accumularsi in alcune regioni del cervello senza poter essere distrutta. Inoltre la proteina alterata può portare addirittura all'alterazione delle proteine prioniche normali prodotte dalle nostre cellule, innescando così, un meccanismo a catena con accumulo di prioni alterati.
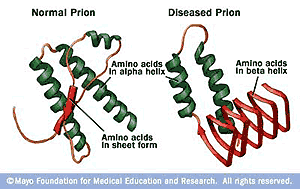
Fig. 5: Differenze strutturali tra proteina prionica normale e mutata
La modificazione che ne deriva è una degenerazione di tipo spongiforme del cervello. Una minore percentuale di individui, possono ammalarsi di questa sindrome qualora avessero nel gene che codifica per la proteina prionica umana, una mutazione che la rendesse simile a quella alterata nel Morbo della Mucca Pazza.
Se per la malattia di Huntinghton e nella Sindrome di Creutzfeld-Jacob le mutazioni di un singolo gene spiegano la maggior parte dei casi, per altre forme patologiche il quadro eziologico appare essere molto più complesso, dovuto sia alla molteplicità di geni che potrebbero essere coinvolti, sia alla loro interazione con “fattori ambientali”.
La cellula, ad esempio, ha evoluto complessi meccanismi che le permettono di eliminare correttamente prodotti di scarto, proteine non funzionanti e componenti cellulari non più necessari. Se uno di questi processi dovesse danneggiarsi, la cellula non sarebbe più in grado di eliminare le scorie, che finirebbero per accumularsi danneggiando l’intero tessuto.
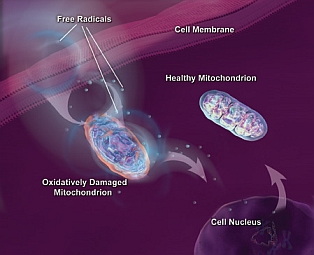 Fig. 6: Meccanismi di danno cellulare indotto da alterazione mitocondriale |
Un altro sistema ritenuto responsabile di molti casi di neurodegenerazione è il mitocondrio. Questo organello, presente in tutti i tipi cellulari del nostro corpo, rappresenta la centrale energetica della cellula, in cui i metaboliti vengono convertiti in energia. Normalmente come prodotti indesiderati di questo processo vengono prodotti i cosiddetti ROS (Radicali Liberi dell’Ossigeno), molto tossici per la cellula, che però vengono eliminati da appositi enzimi o molecole antiossidanti. In condizioni note come stress ossidativo, o danno mitocondriale, i ROS potrebbero essere prodotti in quantità eccessive o la cellula potrebbe non essere in grado di eliminarli correttamente, provocandone il rilascio nel citoplasma con conseguenti danni alle membrane, al DNA o alle proteine stesse.
Anche alterazioni del trasporto assonale![]() possono essere causa, oltre che conseguenza, della neurodegenerazione e possono derivare da malfunzionamento di mitocondri, citoscheletro oppure proteine di trasporto.
possono essere causa, oltre che conseguenza, della neurodegenerazione e possono derivare da malfunzionamento di mitocondri, citoscheletro oppure proteine di trasporto.
Infine, un ruolo fondamentale è determinato dalla cosiddetta “morte cellulare programmata”, a cui la cellula va incontro a un processo simile a un “suicidio”, chiamato in termini scientifici “apoptosi![]() cellulare”. Questo rappresenta un punto in cui convergono tutti i meccanismi di neurodegenerazione, ma che in molti casi può esserne anche un fattore scatenante, e ciò spiega la perdita di neuroni, quindi di materia cerebrale, visibile in pazienti malati tramite tecniche di risonanza magnetica o TAC.
cellulare”. Questo rappresenta un punto in cui convergono tutti i meccanismi di neurodegenerazione, ma che in molti casi può esserne anche un fattore scatenante, e ciò spiega la perdita di neuroni, quindi di materia cerebrale, visibile in pazienti malati tramite tecniche di risonanza magnetica o TAC.
In realtà la ricerca, servendosi anche di modelli animali, sta portando alla luce nuovi possibili scenari in cui una cellula nervosa, ad un certo punto, degenera innescando la malattia. Nascono, così teorie sul ruolo del sistema immunitario, che potrebbe impazzire attaccando le cellule nervose umane, o di sostanze tossiche, come l’esposizione a metalli pesanti, traumi cranici e così via.
Le caratteristiche principali che accomunano tutte le malattie neurodegenerative sono, innanzitutto, il carattere progressivo della malattia, che converge sempre nella morte dei neuroni colpiti. Un altro aspetto comune è senza dubbio l’irreversibilità del processo che, salvo alcune preliminari osservazioni sperimentali, rende il percorso neurodegenerativo irreversibile.
Tuttavia, come già detto, le forme cliniche che possono derivare da tali processi, sono molte e diverse tra loro, a seconda del tessuto colpito dalla malattia e dalle cause che la scatenano.
Molto spesso la malattia neurodegenerativa può essere considerata come un anormale processo di invecchiamento, come dimostrato dal fatto che la maggior parte delle persone colpite sono, per l’appunto, anziane.