La malattia di Huntington
La malattia di Huntington (HD) è una patologia risultante dalla degenerazione geneticamente programmata di neuroni del sistema extrapiramidale e fa parte delle cosiddette sindromi ipercinetiche. Questa degenerazione causa movimenti incontrollati, simili a quelli di una danza (corèa, da qui anche il nome di Corea di Huntington), perdita di facoltà intellettive e disturbi emozionali.
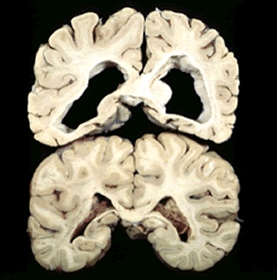 Fig. 1: Degenerazione cerebrale tipica di pazienti affetti da HD. |
Chi viene colpito?
La malattia di Huntington è molto rara, colpisce circa 3-7 individui ogni 100.000 abitanti in Europa, con un’età di esordio intorno a 40-50 anni e decorso clinico che varia da soggetto a soggetto.
Cosa provoca la malattia?
La malattia di Huntington, a differenza delle altre malattie neurodegenerative, è una patologia quasi prettamente familiare, trasmessa, cioè, da genitori a figli tramite una mutazione nel gene che codifica per l’Huntingtina (Htt) secondo una modalità autosomica dominante e penetranza completa.
Come descritto nella prima pagina, tutti i pazienti affetti dalla malattia presentano un numero di ripetizioni della sequenza trinucleotidica CAG in questo gene, superiore a 36, laddove negli individui non affetti questa varia da 11 a 35 volte. La malattia è soggetta anche al fenomeno genetico dell’anticipazione; risulta, infatti comparire tanto più precocemente, quanto maggiore è il numero delle ripetizioni e di solito questo numero tende ad aumentare tra padre e figli per errori nella replicazione del DNA che si verificano durante la spermatogenesi.
In genere, però, la regione diventa instabile e soggetta a rischio di mutazione solo in soggetti che abbiano un numero di triplette superiore a 28; si parla in questo caso di condizione intermedia o pre-patologica
Caratteristiche patologiche
L’Htt è una proteina ampiamente espressa nei neuroni, ma le sue funzioni sono ancora in larga parte sconosciute. Si pensa che possa avere un ruolo protettivo per i neuroni. L’aumento di queste triplette nel gene, che corrispondono all’aumento di aminoacido Glutamina nella sequenza proteica, potrebbe far perdere alla proteina le sue capacità protettive, o probabilmente farne acquisire nuove, tossiche per la cellula.
La malattia è caratterizzata dalla formazione di inclusioni intranucleari e aggregati proteici. È probabile che in un primo momento gli aggregati si formino in seguito ad un meccanismo difensivo della cellula, che sequestrerebbe la proteina mutata, ma col passare del tempo questi aggregati finirebbero per impedire il traffico di molecole nella cellula.
La Malattia di Huntington è, purtroppo, incurabile e gli unici farmaci disponibili possono solo migliorare alcuni sintomi come i movimenti involontari, mentre nulla possono per fermare la progressione della malattia.